Heats of Formation of Medium-Sized Organic Compounds from Contemporary Electronic Structure Methods

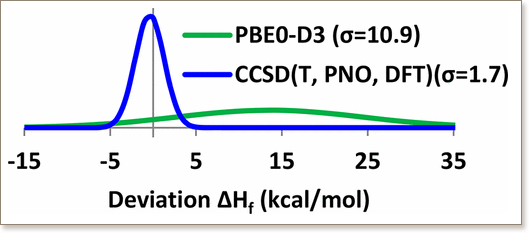
Computational electronic structure calculations are routinely undertaken to predict thermodynamic properties of various species. However, the application of highly accurate wave function theory methods, such as the “gold standard” coupled cluster approach including single, double, and partly triple excitations in perturbative fashion, CCSD(T), to large molecules is limited due to high computational cost. In this work, the promising domain based local pair natural orbital coupled cluster approach, DLPNO–CCSD(T), has been tested to reproduce 113 accurate formation enthalpies of medium-sized molecules (few dozens heavy atoms) important for bio- and combustion chemistry via the reaction based Feller–Peterson–Dixon approach. For comparison, eight density functional theory (B3LYP, B3LYP-D3, PBE0, PBE0-D3, M06, M06-2X, ωB97X-D3, and ωB97M-V) and MP2-based (B2PLYP-D3, PWPB95-D3, B2T-PLYP, B2T-PLYP-D, B2GP-PLYP, DSD-PBEP86-D3, SCS-MP2, and OO-SCS-MP2) methods have been tested. The worst performance has been obtained for the standard hybrid DFT functionals, PBE0 (mean unsigned error (MUE)/mean signed error (MSE) = 9.1/6.0 kcal/mol) and B3LYP (MUE/MSE = 13.5/–13.3 kcal/mol). The influence of an empirical dispersion correction term on these functionals’ performance is not homogeneous: B3LYP performance is improved (B3LYP-D3 (MUE/MSE = 6.0/0.8 kcal/mol)); meanwhile PBE0 performance is worse (PBE0-D3 (MUE/MSE = 14.1/13.6 kcal/mol)). The Minnesota functionals, M06 (MUE/MSE = 3.8/–2.0 kcal/mol) and M06-2X (MUE/MSE = 3.5/3.0 kcal/mol), and recently developed ωB97X-D3 (MUE/MSE = 3.2/0.2 kcal/mol) and ωB97M-V (MUE/MSE = 2.2/1.3 kcal/mol) methods provided significantly better formation enthalpies. Enthalpies of similar quality can also be obtained from some double hybrid methods (B2PLYP-D3 (MUE/MSE = 4.7/2.0 kcal/mol), PWPB95-D3 (MUE/MSE = 4.3/3.2 kcal/mol), B2T-PLYP (MUE/MSE = 4.1/–3.0 kcal/mol), and B2T-PLYP-D (MUE/MSE = 3.3/1.7 kcal/mol)). The two spin component scaled (SCS) MP2 methods resulted in even smaller errors (SCS-MP2 (MUE/MSE = 1.9/1.2 kcal/mol) and OO-SCS-MP2 (MUE/MSE = 1.6/0.1 kcal/mol)). The best performance was found for the frozen core (FC) DLPNO–CCSD(T) method with a MUE/MSE of 1.6/–1.2 kcal/mol. The performance of the DLPNO–CCSD(T) method can be further improved by running the post-SCF calculations on the B3LYP orbitals: the MUE/MSE for the DLPNO–CCSD(T,B3LYP) approximation is 1.2/–0.4 kcal/mol. We recommend the DLPNO–CCSD(T,B3LYP) method for black box applications in thermodynamics of medium-sized organic molecules when the canonical CCSD(T) calculations with basis sets of reasonable quality are prohibitively expensive.
"KAUST shall be a beacon for peace, hope and reconciliation, and shall serve the people of the Kingdom and the world."
King Abdullah bin Abdulaziz Al Saud, 1924 – 2015
Thuwal 23955-6900, Kingdom of Saudi Arabia
© King Abdullah University of Science and Technology. All rights reserved