Theoretical kinetic study of the unimolecular keto–enol tautomerism propen-2-ol ↔ acetone. Pressure effects and implications in the pyrolysis of tert- and 2-butanol

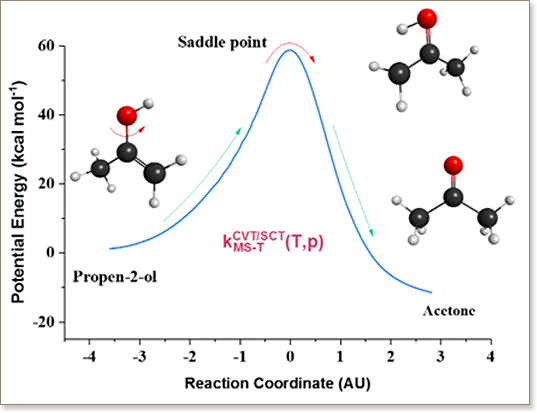
The need for renewable and cleaner sources of energy has made biofuels an interesting alternative to fossil fuels, especially in the case of butanol isomers, with its favorable blend properties and low hygroscopicity. Although C4 alcohols are prospective fuels, some key reactions governing their pyrolysis and combustion have not been adequately studied, leading to incomplete kinetic models. Enols are important intermediates in the combustion of C4 alcohols, as well as in atmospheric processes. Butanol reactions kinetics is poorly understood. Specifically, the unimolecular tautomerism of propen-2-ol ↔ acetone, which is included in butanol combustion kinetic models, is assigned rate parameters based on the tautomerism vinyl alcohol ↔ acetaldehyde as an analogy. In an attempt to update current kinetic models for tert- and 2-butanol, a theoretical kinetic study of the titled reaction was carried out by means of CCSD(T,FULL)/aug-cc-pVTZ//CCSD(T)/6-31+G(d,p) ab initio calculations, with multistructural torsional anharmonicity and variational transition state theory considerations in a wide temperature and pressure range (200-3000 K; 0.1-108 kPa). Results differ from vinyl alcohol ↔ acetaldehyde analogue reaction, which shows lower rate constant values. It was observed that decreasing pressure leads to a decrease in rate constants, describing the expected falloff behavior. Tunneling turned out to be important, especially at low temperatures. Accordingly, pyrolysis simulations in a batch reactor for tert- and 2-butanol with computed rate constants showed important differences in comparison with previous results, such as larger acetone yield and quicker propen-2-ol consumption.
DOI: 10.1021/acs.jpca.8b00836
"KAUST shall be a beacon for peace, hope and reconciliation, and shall serve the people of the Kingdom and the world."
King Abdullah bin Abdulaziz Al Saud, 1924 – 2015
Thuwal 23955-6900, Kingdom of Saudi Arabia
© King Abdullah University of Science and Technology. All rights reserved